Is Legacy Documentation Killing Your Platform Reuse? 🛑
Imagine you’ve developed a world-class injection-pen platform. It’s proven, it’s reliable, and you want to reuse it for a new therapy.
Then, a new EU MDR guidance update lands.
Shortly after, updated expectations from the FDA emerged for combination products. At the same time, global authorities like EMA, PMDA, and Health Canada are tightening requirements for device analytics, software verification, risk data, and lifecycle evidence.
Suddenly, “the same pen” requires a mountain of new paperwork.
For many MedTech leaders, this is where the platforming strategy breaks down.
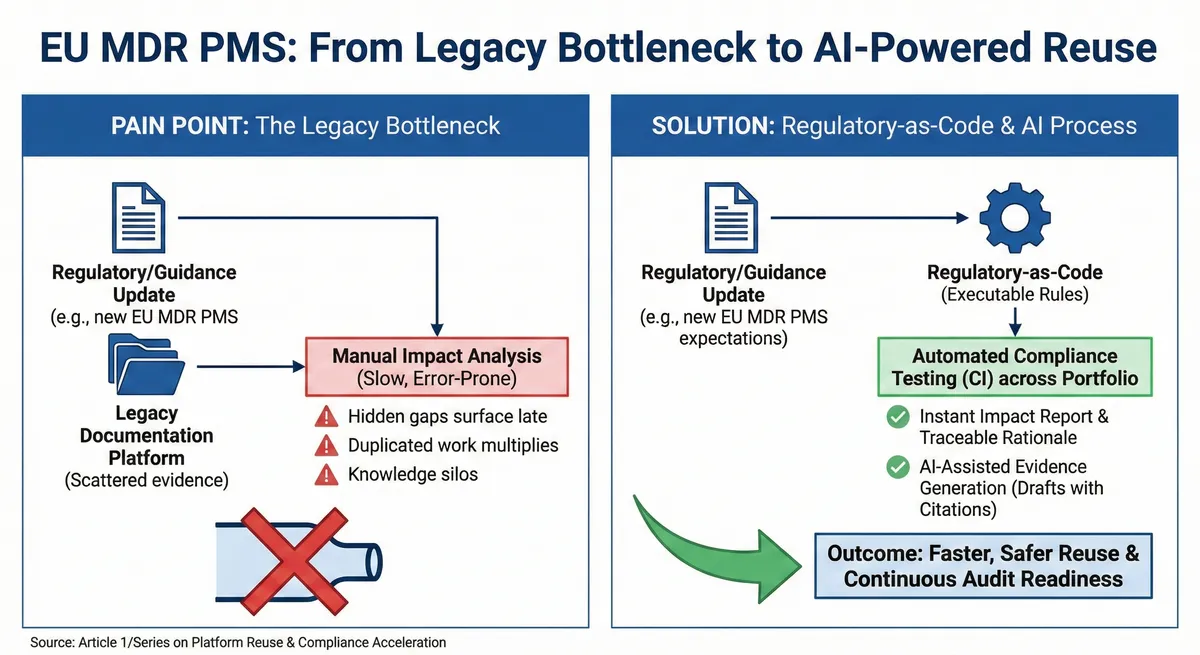
The reuse dream does not fail in engineering, it fails in the documentation. The evidence required to justify reuse is often buried across thousands of legacy PDFs, meaning manual impact analysis becomes slow, expensive, and prone to human error.
The Core Problem: The “Doc-Centric” Bottleneck
Under today’s regulatory expectations, being in compliance is not a checkbox, it is a continuously evolving obligation. When regulations evolve, you are not just updating one SOP. You are often forced to manually hunt through:
- Requirement documentation
- Verification Reports
- PMS Plans (thresholds and indicators)
- Trend detection methodologies
- Clinical evaluations reports
- Risk management files
When you have a platform (like a pre-filled pen) used across several different drugs, a single regulatory shift triggers a massive wave of duplicated manual work.
The Future: Compliance-as-Code & AI
We are moving away from “searching through documents” to “querying data.” Here is how modern platforms are solving the reuse trap:
- Regulatory-as-Code (RaC): Turn complex global requirements into digital, versioned rules. Instead of reading a paragraph, the system “knows” exactly which device families require specific thresholds.
- Automated Compliance Testing: Think of it like “spellcheck” for your entire portfolio. If a guidance update happens, you run a scan. In minutes, the system flags exactly which products and variants are impacted by a new guidance.
- AI-Assisted Drafting: Use AI to extract evidence from legacy files and draft updated technical documentation with direct citations so humans stay in total control.
The Bottom Line
Platform reuse should be a competitive advantage, not a regulatory burden. By digitizing your compliance logic, you move from reactive firefighting to continuous audit-readiness.
Coming up next in this article series:
- Legacy test rationale reuse (automated testing)
- Tech file comparability & modular documentation
- AI-assisted evidence generation for submissions and audits
How is your team currently mapping regulatory changes to your device portfolio?